Data Science in Life Sciences

Team Leader Data Science in Life Sciences: Prof. Dr. Abdullah Kahraman
Computational identification of new cancer and disease mechanisms for superior clinical diagnostics and therapeutics.
Continuous new developments in DNA sequencing and mass spectrometry have opened up the possibility of probing the molecular landscape of single and bulk cells on an unprecedented scale.
For example, The Cancer Genome Atlas (TCGA) and the International Cancer Genomics Consortium (ICGC) have applied Next Generation Sequencing (NGS) technologies to screen the tumor mutational landscape of more than 85,000 cancer patients, generating over 3 petabytes of data. Using this large dataset, multi-institutional research teams have identified hundreds of cancer driver genes, which can drive cancerous growth in normal cells upon mutations. With this knowledge, pharma companies have started developing novel targeted and immuno- therapies for precision medicine.
Our research is in alignment with the aforementioned international and multi-institutional research programs for precision medicine. We have extensive experience in the development of novel Omics-based diagnostics algorithms and the analysis of non-coding mutations in cancer patients. We focus in particular on the detection of cancer-specific alternative splicing events for diagnostic purposes. For our investigations, we integrate whole-genome Next-Generation-Sequencing data, RNA sequencing data, Mass-Spectrometry based proteomics data, experimental X-ray protein structures, and protein interaction network data.
Cancer-specific Alternative Splicing Events
Alternative RNA splicing is a regulatory cellular mechanism to create multiple mRNA molecules from the same gene and is often disrupted in diseases. We could show in the international Pan-Cancer Analysis of Whole Genomes (PCAWG) study that such disruptions in alternative splicing patterns are widespread in cancer. By developing new computer algorithms, machine learning models, and databases (www.caniso.net) we aim to further understand the origins and consequences of such alternative splicing disruption and work towards developing new splicing biomarkers for superior diagnostic, treatment, and medication.
NGS Data Interpretation
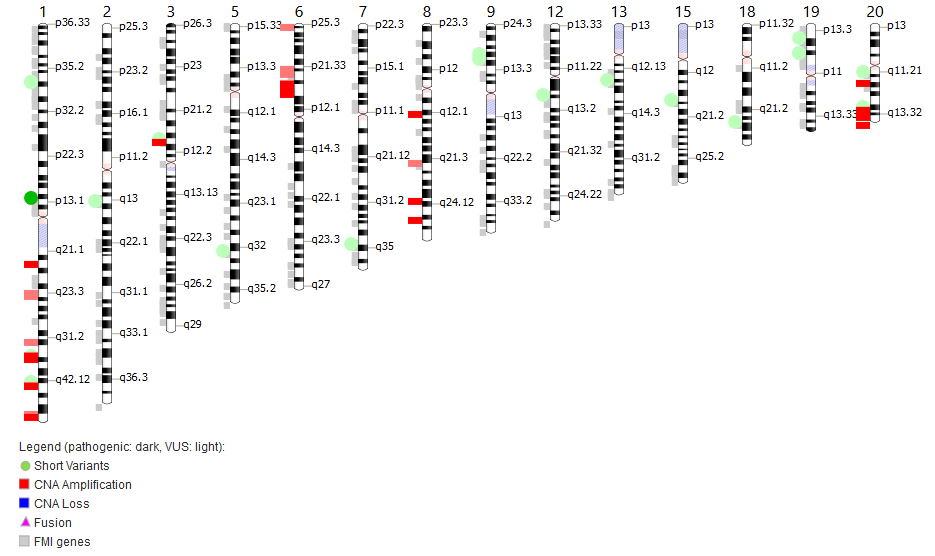
Molecular tumor boards are interdisciplinary meetings in hospitals, where oncologists, pathologists, bioinformaticians, and molecular biologists meet up to discuss Next Generation Sequencing (NGS) results of tumor biopsies. Since costs for NGS assays are dropping, hospitals have started to screen larger regions of cancer genomes for actionable mutations. The increasing size of the assays, however, increases the complexity of NGS results. To support the interpretation and streamline the discussions at Molecular Tumorboards, our group has developed the MTPpilot software (www.MTPpilot.org) to support the interpretation of complex NGS results at molecular tumor boards. We are continuously working on improving the software, adding new functionalities, and creating novel visualization tools.
Data Driven Molecular Modelling
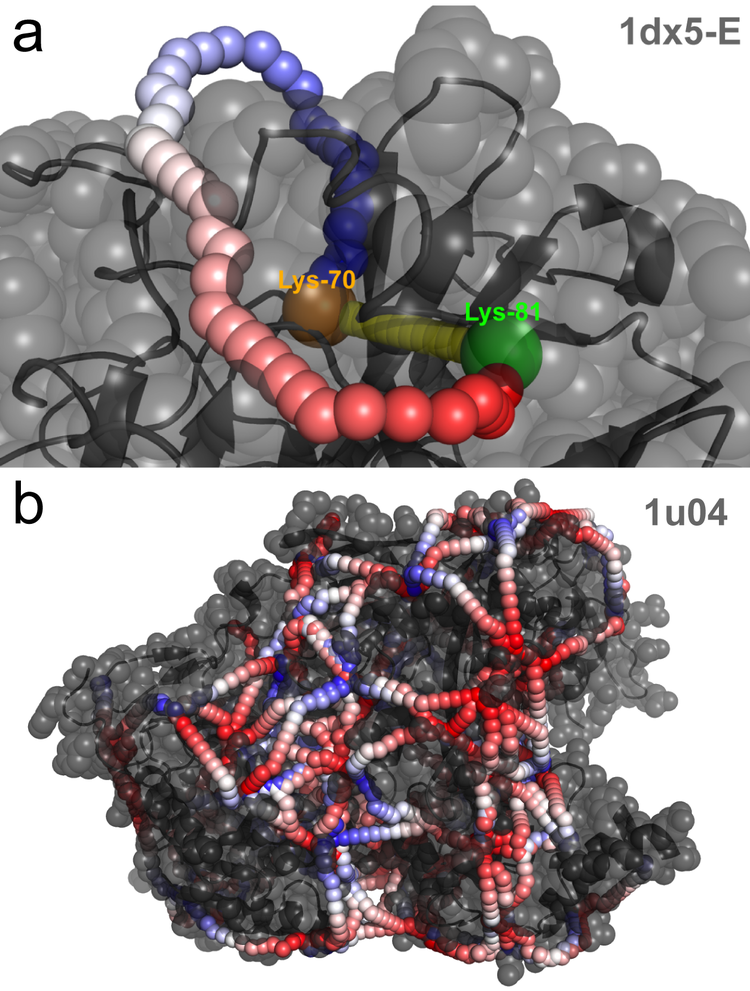
Cancer cells have many deregulated protein complexes. Traditionally, these multimeric protein complexes have been studied by X-ray crystallography and cryo-Electron-Microscopy (cryo-EM). Recently, structural proteomics techniques like chemical Cross-linking Mass Spectrometry (CX-MS) and Limited Proteolysis coupled to targeted Mass-spectrometry (LiP-SRM) emerged as powerful complementary techniques. By integrating data from these new techniques with data-driven modeling via ROSETTA and AlphaFold, we aim to predict the structure of protein isoforms and large protein complexes in cancer cells. The structural information will help us to understand the functional impact of mutations and protein isoforms on cellular complexes and pathways.

PI
We are interested in understanding and targeting the impact of clinically relevant alternative splicing disruptions in cancer, aging, and disease by integrating omics, protein design, and AI/ML in a clinical setting to identify and develop early cancer and disease biomarkers and therapies.

Postdoc
I am a postdoctoral researcher at the FHNW Data Science in Life Sciences Lab. As an expert at the intersection of generative AI and structural biology, I specialize in data-intensive and high-performance computing.
My research focuses on cancer genomics, with a particular interest in investigating aberrant alternative splicing to discover novel therapeutic targets. I employ state-of-the-art machine learning and generative AI techniques, such as protein structure prediction and de novo design, alongside classical structural biology methods. Through the integration of computational chemistry and molecular modelling, I aim to accelerate rational drug discovery and target validation.

Postdoc
I am a postdoctoral researcher specialized in developing probabilistic cancer models, particularly Bayesian and Markov approaches combined with multi-omics data. During my PhD, I developed wet-lab supporting workflows by integrating computational methods with experimental observations to understand stress-induced metastatic phenotypes. I also worked as a computational consultant on industry projects focused on therapeutic target discovery in hot and cold tumors. My current research focuses on understanding how aberrant alternative splicing shapes the tumor microenvironment and detecting targetable mechanisms for translational oncology projects.

PhD Student
I am a Ph.D. student with a focus on applying advanced computational methods, including Machine Learning (ML) and Deep Learning, to complex biological problems. My current research, conducted at the FHNW Data Science in Life Sciences Lab, focuses on cancer biology and personalized medicine, specifically by investigating splice sites to inform individualized drug strategies. Leveraging my expertise in bioinformatics, computational data analysis, and genomics, I am committed to driving innovative research at the intersection of structural and computational biology.

As a research intern at the Kahraman lab, my work currently focuses on cancer genomics. I am investigating the splicing landscape of different gene fusions observed in TFE3-rearranged renal cell carcinoma. Alongside my role in the group, I am studying the Data Science in Life Sciences MSc at FHNW.

Remote Research Intern
As a Senior Bachelor student in Molecular Biology and Genetics, I work as a research intern in the Kahraman Lab, focusing on computational cancer immunology. My research involves analyzing tumor RNA-seq data to identify mutation and splicing-derived neoepitopes with therapeutic potential. I continue developing my skills in immunogenomics and cancer bioinformatics.
CanIsoNet: Predicting the pathogenic impact of isoform switches using isoform-specific interaction networks
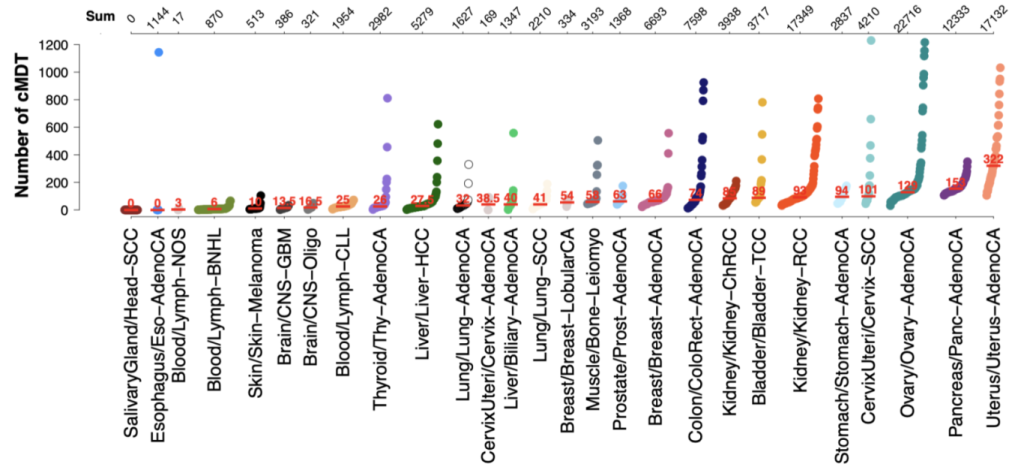
Alternative splicing regulation is often disturbed in various cancers leading to cancer-specific switches in the Most Dominant Transcripts (cMDT). To understand how these switches drive oncogenesis, we have developed an isoform-specific protein interaction network that can be used to study the impact of isoform switches in cancer samples. The highlighted common and distinct patterns of alternative splicing deregulations constitute new avenues for novel therapeutic targets in the fight against cancer.
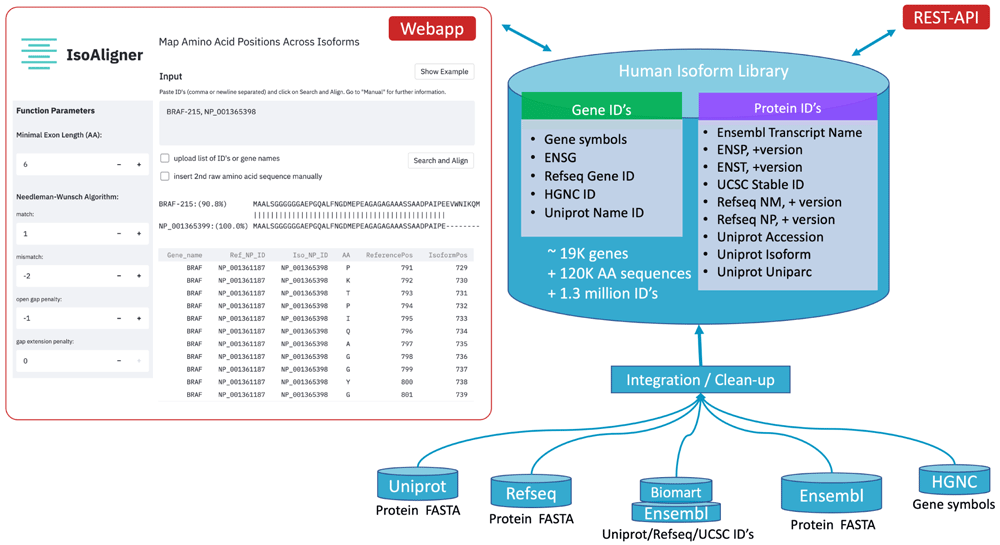
IsoAligner: Map Amino Acid Positions Across Protein Isoforms
Aligning protein isoform sequences is often performed in cancer diagnostics to homogenise mutation annotations from different diagnostic assays. However, most alignment tools are fitted for homologous sequences, leading often to alignments of non-identical exonic regions. To address this problem, we have implemented an interactive alignment webservice called IsoAligner for exact mapping of exonic protein subsequences.
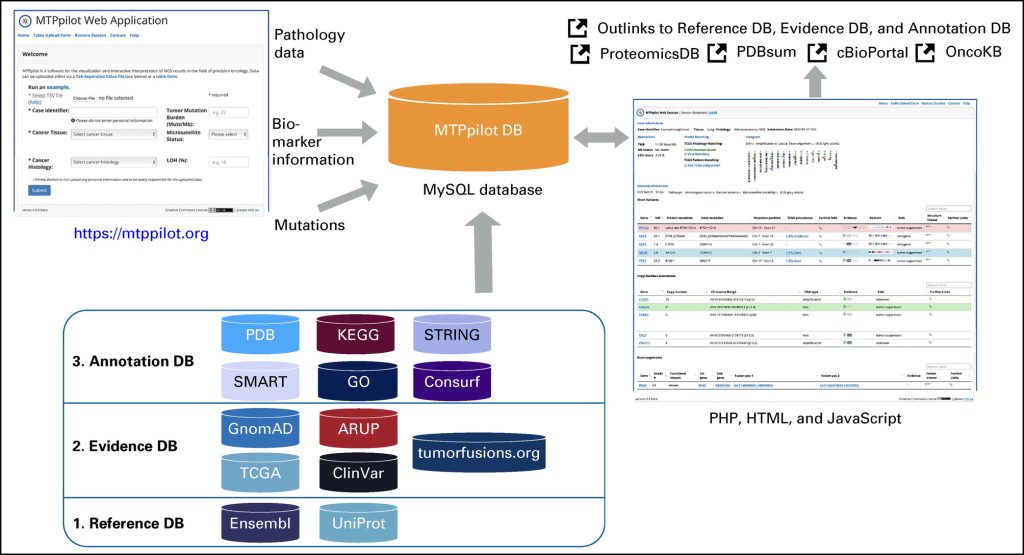
MTPpilot: Visualization and interactive interpretation of NGS results in the field of precision oncology
Comprehensive targeted next-generation sequencing (NGS) panels are routinely used in modern molecular cancer diagnostics. In molecular tumor boards, the detected genomic alterations are often discussed to decide the next treatment options for patients with cancer. With the increasing size and complexity of NGS panels, the discussion of these results becomes increasingly complex, especially if they are reported in a text-based form, as it is the standard in current molecular pathology.
We have developed the Molecular Tumor Profiling pilot (MTPpilot) web service using HTML, PHP, JavaScript, and MySQL to support the clinical discussion of NGS results at molecular tumor boards.
MTPpilot integrates various public genome, network, and cancer mutation databases with interactive visualization tools to assess the functional impact of mutations and support clinical decision-making at tumor boards.
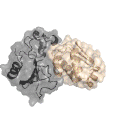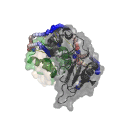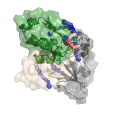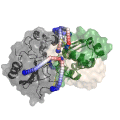
Xwalk: Prediction, Validation and Visualisation of Chemical Cross-Link Data.
Chemical cross-linking of proteins or protein complexes and the mass spectrometry-based localization of the cross-linked amino acids is a powerful method for generating distance restraints on the substrate’s topology. Xwalk was written to predict and validate these cross-links on existing protein structures. Xwalk calculates and displays non-linear distances between chemically cross-linked amino acids on protein surfaces while mimicking the flexibility and non-linearity of cross-linker molecules. It returns a Solvent Accessible Surface Distance, which corresponds to the length of the shortest path between two amino acids, where the path leads through solvent-occupied space without penetrating the protein surface.
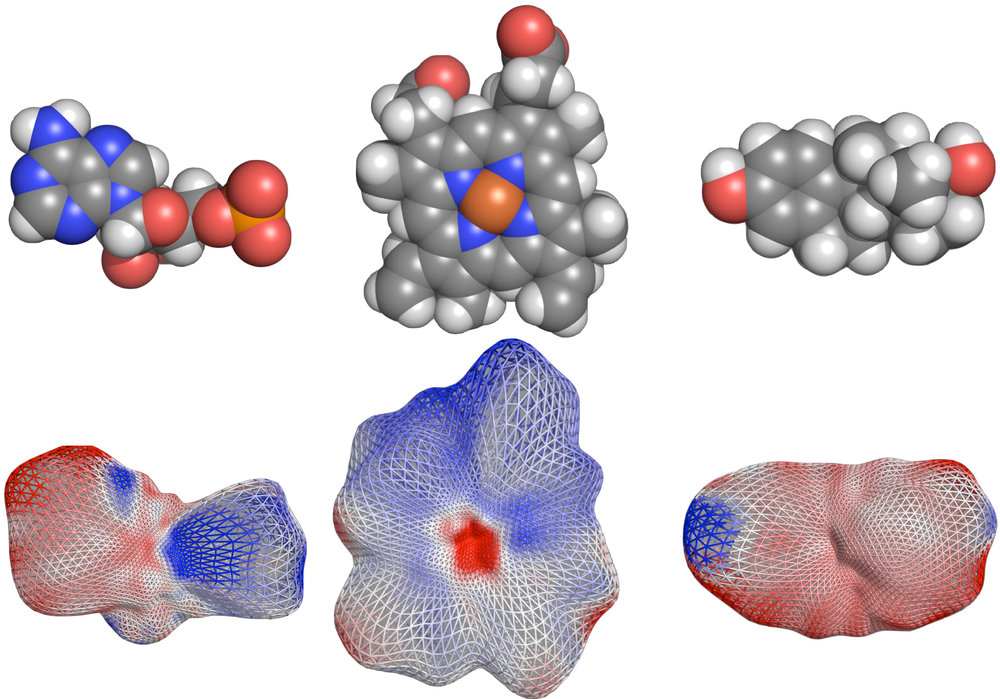
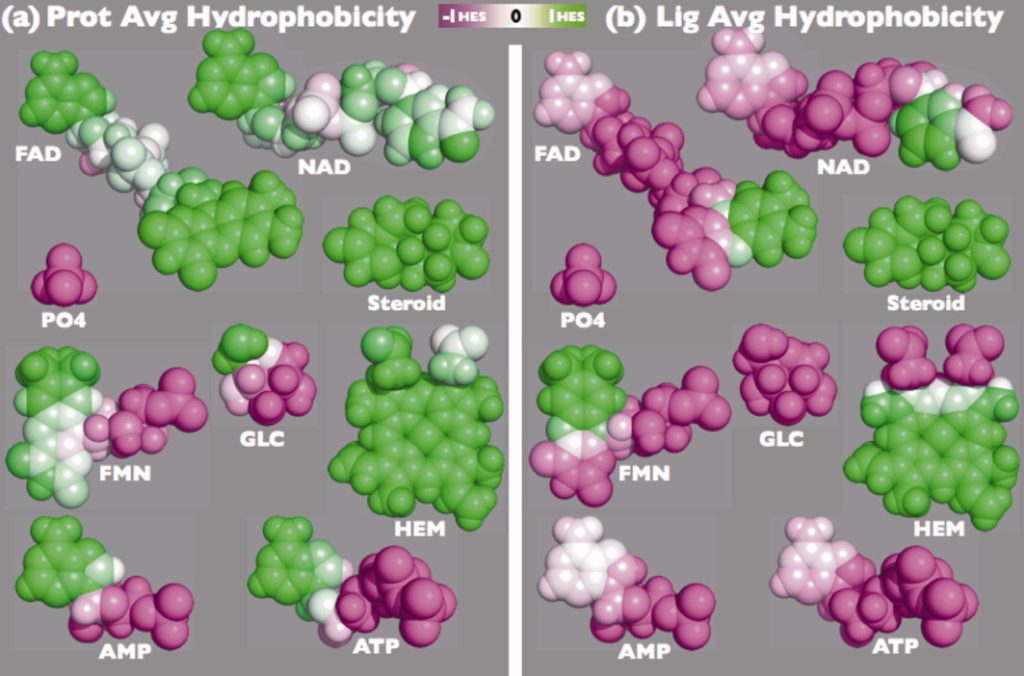
CleftXplorer: Geometrical and Physicochemical Analysis and Comparison of Protein Binding Pockets and Ligands.
Compare protein binding pockets with each other or small molecules using spherical harmonics. Analyze the electrostatic potential, hydrophobicity, hydrogen bond pattern, and van der Waals forces in protein binding pockets and small molecules. Assess the complementarity between proteins and small molecules.
IsoNet: Isoform-specific Protein Interaction Network
The first transcript isoform-specific interaction network database was created using a smart combination of the databases STRING, 3did and ENSEMBL. A detailed description of how it was generated, can be found in the methods section of our recent paper. The entire network is available in Table S2.
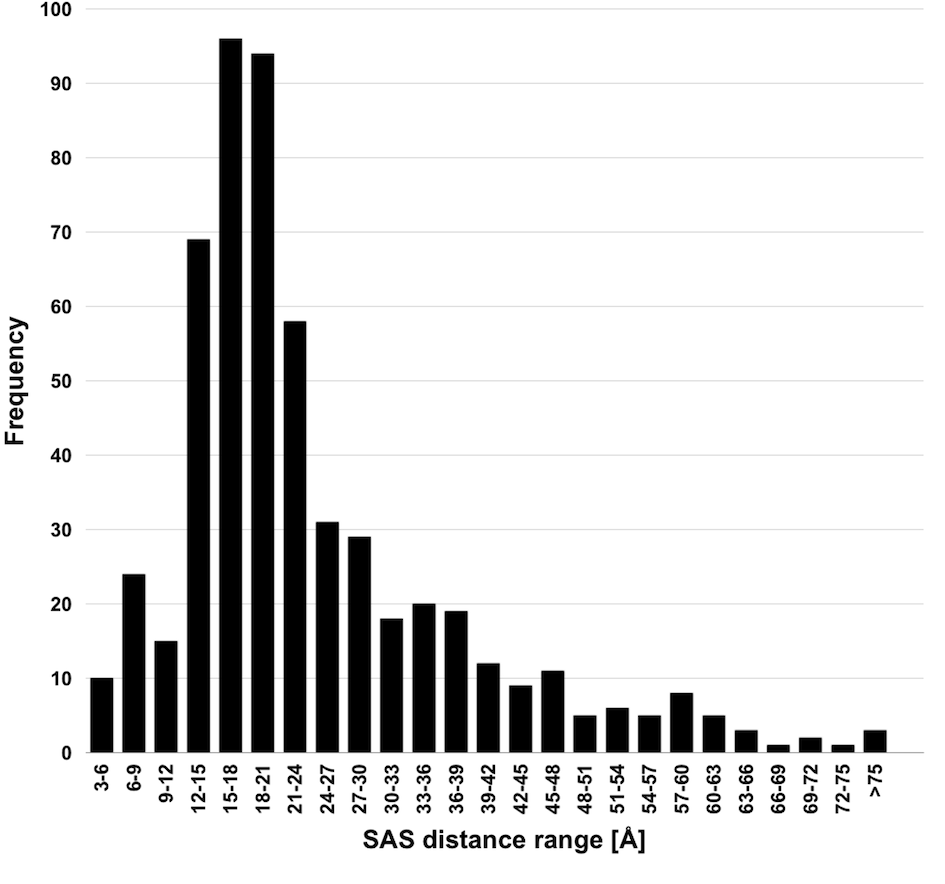
XLdb, A chemical Cross-Link Database
Chemical cross-linking combined with mass-spectrometry (XL-MS) data from 14 different publications were collected. The database encompasses a total number of 506 intra-protein and 62 inter-protein cross-links that were all generated with the disuccinimidyl suberate (DSS) or bis(sulfosuccinimidyl) suberate (BS3) cross-linker reagent. Experimental structural data in the Protein Data Bank (PDB) exists for all cross-linked proteins. The structural data permits the mapping of each chemical cross-link on a protein structure, which is essential for distance measurements of XL-MS data (see Table S1).
Kahraman Dataset: Dataset of 100 protein binding pockets spread over nine ligand sets
Matching small molecules to protein binding sites is an important problem in protein docking and protein function prediction. To support method developments in this field, we created a dataset with 100 non-homologous protein binding sites that bind one of nine ligand of different size and shape, including phosphate as the smallest and most rigid molecule to ATP as flexible and middle-sized molecule up to FAD as the biggest and most flexible molecule. Since in a non-homologous data, one can expect that proteins have evolved different strategies for binding the same ligand, the conformation and physicochemistry of binding sites varies between the proteins. The dataset is available in Table 1.
01.12.2025: Abdullah just got an affiliation in the group of Prof. Dr. Viktor Kölzer as a Topic Lead in Clinical Bioinformatics at the University Hospital Basel. The new affiliation will foster the collaboration between HLS and USB and allow the Kahramanlab to continue its clinically relevant cancer research in molecular diagnostics and Molecular Tumor Boards. Stay tuned for news on our research outputs.
01.12.2025: Welcome, Yavuzhan, our new Ph.D. student. We can’t wait to do great science with you.
02.10.2025: The second installment of our “Life Science Industry Meets Data Science” has just kicked off. It’s gonna be an awesome day full of talks on Generative AI, Bioprocessing, Personalized Medicine and Health, and a collective intelligence session to help Genorare B.V. shape their business case. See here for the agenda.
17.09.2025: We did it! Our InnoSuisse Flagship proposal, “NAIPO: National AI Initiative for Precision Oncology,” has been accepted. Stay tuned for more news soon. This is our LinkedIn post about our success.
08.09.2025: SIB’s BC2 2025 conference has just started. Three highly engaging, motivating, and inspiring days are awaiting us. We are thankful for having had the opportunity to co-organize the event. Find the agenda and more here.
28.08.2025: Proud to have co-organized the 5th Clinic Meets Data Science Symposium at the University Hospital Basel. The symposium again demonstrated how cross-disciplinary collaboration is shaping the future of medicine – from AI-assisted diagnostics to integrative omics, from digital twins to patient-centered data platforms. Find key takeaways here.
05.09.2025: A great welcome to Ermin Taric, our new BSc student. Ermin will work on AI-generated isoform-specific protein interaction networks. Looking very much forward to working with you!
19.08.2025: A warm welcome to Daniela, our new Master Research Intern. We are thrilled to have you with us. Looking forward doing great science with you.
10.07.2025: We had an amazing “1st Swiss Symposium on Protein Design for Academia and Industry” with a leading Swiss expert on protein design, hands-on afternoon sessions, and an audience packed with great scientists from academia and industry. Find more information here.
07.07.2025: Welcome to our Turkish summer students, Sena and Eymen. It’s great to have you with us. We look forward to doing some great bioinformatics work with both of you.
14.04.2025: Our new paper on using single-cell long-read sequencing to understand the landscape of known and novel transcripts in ccRCC is out now. Check it out here.
07.04.2025: Welcome, Felix Frank, our new BSc student, who will work on conformational changes induced by disease-causing mutations and isoforms using AlphaFold and Molecular Dynamics.
01.03.2025: Welcome, Jakob, to our group. Can’t wait to work with you on exciting science.
23.06.2024: It’s an honor to host my PhD mother, Prof Dame Janet Thornton, at HLS. She is one of the founders of the Bioinformatics field, directed the European Bioinformatics Institute in Cambridge/UK for 14 years, won numerous awards and prices, was appointed Dame Commander of the British Empire, is a fellow of the Royal Society, EMBO, US National Academy of Sciences, FMedSci, FRSC, is author of one of the most cited papers in history … the list goes on. So do not miss the opportunity to see her and listen to her take on the importance of bioinformatics for life science research. Find all the information here.
04.05.2023: Join us for the 3rd installment of our symposium “Clinics Meets Data Science 2023”. For free registration and more information, follow this link.
17.04.2023: Welcome Simon Bernèche, who is joining our group as a computational research scientists. Looking forward to doing science together.
17.04.2023: Our database on disease-specific alternative splicing events has just been published in Bioinformatics Advances: CanIsoNet: a database to study the functional impact of isoform switching events in diseases.
14.02.2023: Our collaboration with the Pauli lab @ USZ on “Unravelling homologous recombination repair deficiency and therapeutic opportunities in soft tissue and bone sarcoma” has just been published in EMBO Mol Med. Read the paper here.
06.02.2023: We were granted a Research Grant from the School for Life Sciences to pursue a project on Quantum accelerated prediction of isoform-specific drug interactions with ROSETTA.
02.02.2023: We are co-organizing a practical workshop for (foodborne) pathogen detection from (direct Nanopore) sequencing data using Galaxy in collaboration with the Galaxy Team at the University of Freiburg and Bioloytix here at our campus in Muttenz.
31.01.2023: We are attending the Biotechnet Meet-Up 2023 event in Sion, Switzerland, where we will introduce our new Biotechnet Data Science in Life Science platform that we founded together with Alexandre Kuhn (HEVS), Lukas Neutsch (ZHAW) and Moritz Kirschmann (CSEM). Stayed tuned for our future networking events and services.
30.01.2023: Our School for Lifesciences is organizing a Symposium on Digital Methods in Life Sciences, highlighting the Past, Present, and Future of digital methods developed at the School. It’s an exciting program with 18 speakers, a keynote given by Nicholas Kelley, the Director of Data Science & A.I. Innovation and Chief Digital Office at Novartis, and a lot of networking possibilities. We are looking forward to the event.
03.11.2022: Our new lab website @FHNW is live
43. Heesen, P., Christ, S.M., Ciobanu-Caraus, O., Kahraman, A. et al. (2025). Clinical prognostic models for sarcomas: a systematic review and critical appraisal of development and validation studies. Diagn Progn Res 9, 7.
Dlamini, Z., Ladomery, M.R., and Kahraman, A. (2024). Editorial: The RNA revolution and cancer.
Front. Endocrinol. 15, 1422599.
42. Karakulak, T., Bolck, H.A., Zajac, N., Bratus-Neuenschwander, A., Zhang, Q., Qi, W., Oltra, T.C., Rehrauer, H., Mering, C. von, Moch, H., Kahraman, A. (2024). Heterogeneous and Novel Transcript Expression in Single Cells of Patient-Derived ccRCC Organoids. Genome Res., 35, 698–711.
41. Zajac, N., Zhang, Q., Bratus-Neuschwander, A., Qi, W., Bolck, H.A., Karakulak, T., Oltra, T.C., Moch, H., Kahraman, A., and Rehrauer, H. (2025). Comparison of Single-cell Long-read and Short-read Transcriptome Sequencing of Patient-derived Organoid Cells of ccRCC: Quality Evaluation of the MAS-ISO-seq Approach, NAR Genomics and Bioinformatics, Volume 7, Issue 3, lqaf089.
40. Angori, S., Banaei-Esfahani, A., Mühlbauer, K., Bolck, H.A., Kahraman, A., Karakulak, T., Poyet, C., Feodoroff, M., Potdar, S., Kallioniemi, O., Pietiäinen, V., Schraml, P., Moch, H (2023). Ex Vivo Drug Testing in Patient-derived Papillary Renal Cancer Cells Reveals EGFR and the BCL2 Family as Therapeutic Targets. Eur Urol Focus. Sep;9 (5):751-759.
39. Karakulak, T., Szklarczyk, D., Moch, H., von Mering, C., Kahraman, A. (2023). CanIsoNet: A Database to Study the Functional Impact of Isoform Switching Events in Cancer. Bioinformatics Advances, Volume 3, Issue 1, 2023, vbad050.
39. Akhoundova, D., Hussung, S., Sivakumar, S., Töpfer, A., Rechsteiner, M., Kahraman, A., et al. (2022) ROS1 genomic rearrangements are rare actionable drivers in microsatellite stable colorectal cancer. Int J Cancer. 2022;151:2161–71.
38. Kahraman, A., Buljan, M. and Vitting-Seerup, K., (2022). Alternative Splicing in Health and Disease. Frontiers in Molecular Biosciences, 9.
37. Kahraman, A., Arnold, F. M., Hanimann, J., Nowak, M., Pauli, C., Britschgi, C., Moch, H., & Zoche, M. (2022). MTPpilot: An Interactive Software for Visualization of Next-Generation Sequencing Results in Molecular Tumor Boards. JCO clinical cancer informatics, 6, e2200032.
36. Hanimann, J., Moch, H., Zoche, M. and Kahraman, A. (2022). IsoAligner: dynamic mapping of amino acid positions across protein isoforms. F1000Research 2022, 11:382
35. Degirmenci, B., Dincer, C., Demirel, H.C., Berkova, L., Moor, A.E., Kahraman, A., Hausmann, G., Aguet, M., Tuncbag, N., Valenta, T. and Basler, K., (2021). Epithelial Wnt secretion drives the progression of inflammation-induced colon carcinoma in murine model. Iscience, 24(12), p.103369.
34. Karakulak, T., Moch, H., von Mering, C., Kahraman, A. (2021). Probing Isoform Switching Events In Various Cancer Types: Lessons From Pan-Cancer Studies. Frontiers in Molecular Biosciences, 23 November 2021.
33. Sobottka, B., Nienhold, R., Nowak, M., Hench, J., Haeuptle, P., Frank, A., Sachs, M., Kahraman, A., et al. (2021). Integrated Analysis of Immunotherapy Treated Clear Cell Renal Cell Carcinomas: An Exploratory Study, Journal of Immunotherapy, 2022 Jan 1;45(1):35-42.
32. Ak, M., Kahraman, A., Arnold, F.M., Turko, P., Levesque, M.P., Zoche, M., Ramelyte, E. and Dummer, R., (2021). Clinicopathological and Genomic Profiles of Atypical Fibroxanthoma and Pleomorphic Dermal Sarcoma Identify Overlapping Signatures with a High Mutational Burden. Genes, 12(7), 974.
31. Irmisch, A., Bonilla, X., Chevrier, S., Lehmann, K. V., Singer, F., Toussaint, N. C., … & Levesque, M. P., (2021). The Tumor Profiler Study: integrated, multi-omic, functional tumor profiling for clinical decision support. Cancer Cell, 39(3), 288-293.
30. Kahraman, A., Karakulak, T., Szklarczyk, D., Mering, von, C. (2020). Pathogenic impact of transcript isoform switching in 1,209 cancer samples covering 27 cancer types using an isoform-specific interaction network. Sci. Rep. 10, 1–15.
29. Hameister, E., Stolz, S.M., Fuhrer, Y., Thienemann F, Schaer, D.J., Nemeth, J., Schuepbach, R.A., Goede, J., Reinhart, S., Schmidt, A., Kahraman, A., Schmid, M., Moch, H., Zoche, M., Manz, M.G., Balabanov, S., Boettcher, S. (2021), Clonal Hematopoiesis in Hospitalized Elderly Patients With COVID-19. HemaSphere 4, e453 (2020).
28. Reyna, M. A., Haan, D., Paczkowska, M., Verbeke, L. P. C., Vazquez, M., Kahraman, A., et al. (2020). Pathway and network analysis of more than 2500 whole cancer genomes. Nature Communications, 11(1), 729.
27. Rheinbay, E., Nielsen, M. M., Abascal, F., Wala, J. A., Shapira, O., Tiao, G., et al. (2020). Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature, 578(7793), 102–111.
26. ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. (2020). Pan-cancer analysis of whole genomes. Nature, 578(7793), 82–93.
25. Rüschoff, J. H., Gradhand, E., Precision, A. K. J. (2019). STRN-ALK Rearranged Malignant Peritoneal Mesothelioma With Dramatic Response Following Ceritinib Treatment. JCO Precision Oncology.
24. Bolck, H. A., Corrò, C., Kahraman, A., von Teichman, A., Toussaint, N. C., Kuipers, J., … & Moch, H. (2021). Tracing clonal dynamics reveals that two-and three-dimensional patient-derived cell models capture tumor heterogeneity of clear cell renal cell carcinoma. European Urology Focus 7(1), 152-162..
23. Schopper, S., Kahraman, A., Leuenberger, P., Feng, Y., Piazza, I., Müller, O., Boersema, P. J., & Picotti, P. (2017). Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry. Nature Protocols, 12(11), 2391–2410.
22. Wang X., Cimermancic P., Yu C., Schweitzer A., Chopra N., Engel J.L., Greenberg C.H., Huzzah A.S., Beck F., Sakata E., Yang Y., Novitsky E.J., Leitner A., Nanni P., Kahraman A., Guo X., Dixon J.E., Rychnovsky S.D., Aebersold R., Baumeister W., Sali A., Huang L. (2017). Molecular Details Underlying Dynamic Structures and Regulation of the Human 26S Proteasome. Molecular & Cellular Proteomics : MCP, 16(5), 840–854.
21. Leuenberger, P., Ganscha, S., Kahraman, A., Cappelletti, V., Boersema, P.J., von Mering, C., Claassen, M., Picotti, P. (2017). Cell-wide analysis of protein thermal unfolding reveals determinants of thermostability. Science 355, 812, eaai7825.
20. SIB Swiss Institute of Bioinformatics Members. (2016) The SIB Swiss Institute of Bioinformatics’ resources: focus on curated databases. Nucleic Acids Res 44, D27–D37.
19. Grimm, M., Zimniak, T., Kahraman, A., Herzog, F. (2015). xVis: a webserver for the schematic visualization and interpretation of crosslink-derived spatial restraints, Nucleic Acids Res. 43, W362–9.
18. Valleliana, F., Garcia-Rubiod, I., Pugliaa, M., Kahraman, A., Deuel, J.W., Engelsberger, W.R., Mason, R.P., Buehlerg, P.W., Schaer, D.J. (2015). Spin trapping combined with quantitative mass spectrometry defines free radical redistribution within the oxidized hemoglobin:haptoglobin complex. Free Radic. Biol. Med. 85, 259–268.
17. Boersema, P., Kahraman, A., Picotti, P. (2015). Proteomics beyond large-scale protein expression analysis. Current Opinion in Biotechnology 34, 162-170.
16. Robinson, M.D., Kahraman, A. , Law, C.W., Lindsay, H., Nowicka, M., Weber, L.M., Zhou, X. (2014). Statistical methods for detecting differentially methylated loci and regions. Front Genet. (5) 324.
15. Feng, Y.*, De Franceschi, G.*, Kahraman, A.*, Soste, M., Melnik, A., Boersema, P., Polverino de Laureto, P., Nikoaev, Y., Oliveira, A.P., Picotti, P. (2014). Global analysis of protein structural changes in complex proteomes. Nature Biotech 32, 1036–1044.
14. Merkley, E.D., Rysavy, S., Kahraman, A., Hafen, R.P., Daggett, V. and Adkins, J.N. (2014). Distance restraints from cross-linking mass spectrometry: Mining a molecular dynamics simulation database to evaluate lysine-lysine distances Protein Science 23 (6), 747-759.
13. Kahraman, A.*, Herzog, F.*, Leitner, A., Rosenberger, G., Aebersold, R., Malmström, L. (2013). Cross-Link Guided Molecular Modeling with ROSETTA. PLoS ONE 8(9): e73411.
12. Herzog, F.*, Kahraman, A.*, Bohringer, D.*, Mak, R., Bracher, A., Walzthoeni, T., Leitner, A., Beck, M., Hartl, F. U., Ban, N, Malmstroem, L., Aebersold, R. (2012). Structural probing of a protein phosphatase 2A network by chemical cross-linking and mass spectrometry. Science 337, 1348–1352.
11. Kahraman, A., Malmström, L., Aebersold, R. (2011). Xwalk: Computing and Visualizing Distances in Cross-linking Experiments. Bioinformatics 27, 2163-2164.
10. Leitner, A., Kahraman, A.*, , Walzthoeni, T.*, Herzog, F., Rinner, O., Beck, M. and Aebersold, R. (2010). Probing native protein structures by chemical cross-linking, mass spectrometry and bioinformatics. Mol Cell Proteomics 9, 1634-1649.
9. Smith, L., Kahraman, A., Thornton, J. M. (2010). Heme proteins – diversity in structural characteristics, function and folding. Proteins 78, 2349-68, PMID: 20544970.
8. Kahraman, A., Morris, R. J., Laskowski, R.M., Favia, A.D., Thornton, J. M. (2010). On the diversity of physicochemical environments experienced by identical ligands in binding pockets of unrelated proteins. Proteins 78, 1120-36, PMID: 19927322.
7. Kahraman, A., Thornton, J. M. (2008). Methods for the analysis of enzyme binding site. Computational Structural Biology: Methods and Applications, Editors: Torsten Schwede, Manuel C. Peitsch. (Amazon).
6. Kahraman, A., Morris, R. J., Laskowski, R. A., Thornton, J. M. (2007). Variation of geometrical and physicochemical properties in protein binding pockets and their ligands. BMC Bioinformatics 8, S1.
5. Kahraman, A., Morris, R. J., Laskowski, R. A., Thornton, J. M. (2007). Shape variation in protein binding pockets and their ligands. J Mol Biol 368, 283-301, PMID: 17337005.
4. Morris, R. J., Kahraman, A., Funkhouser, T., Stockwell, G., Glaser, F., Laskowski, R., Thornton, J. M. (2005). Binding pocket shape analysis for protein function prediction. Quantitative Biology, Shape Analysis, and Wavelets, Leeds University Press, Leeds, 91–94.
3. Morris, R. J., Kahraman, A., Thornton, J. M. (2005). Binding Pocket Shape Analysis for Protein Function Prediction. Acta Crystallographica Section A 61, C156–157.
2. Morris, R. J., Najmanovich, R. J., Kahraman, A., Thornton, J. M. (2005). Real spherical harmonic expansion coefficients as 3D shape descriptors for protein binding pocket and ligand comparisons. Bioinformatics 21, 2347-55, PMID: 15728116.
1. Kahraman, A., Avramov, A., Nashev. L., Popov, D., Ternes, R., Pohlenz, H.D., and Weiss, B. (2005). PhenomicDB: a multi-species genotype/phenotype database for comparative phenomics. Bioinformatics 21, 418-420, PMID: 15374875.

